二級結構
二級結構(英語:Secondary structure)在生物化學及結構生物學中,是指一個生物大分子,如蛋白質及核酸(DNA或RNA),局部區段的三維通式。然而它並不描述任何特定的原子位置(在三級結構中描述)。

二級結構是由生物大分子在原子解像度結構中所觀察到的氫鍵來定義的。蛋白質的二級結構通常是以主鏈中氨基之間的氫鍵模式來定義〈與主鏈-側鏈間以及側鏈-側鏈間的氫鍵無關〉,亦即DSSP的定義。[1]而核酸的二級結構是以鹼基之間的氫鍵來定義。
在二級結構中,特定的氫鍵模式往往伴隨着其他一些結構特徵;但如果只考慮這些結構特徵而忽略氫鍵本身,則會導致所定義的二級結構不準確。例如,蛋白質的螺旋中的殘基都分佈在拉氏圖(以主鏈二面角為坐標)的特定區域,因此二面角位於這一區域的殘基都會被認為參與形成「螺旋」,而不論它是否真正的存在對應氫鍵。其他稍微不準確的定義多是應用曲線微分幾何的觀念,如曲率及扭量。也有一些結構生物學家以肉眼觀察通過軟件顯示的蛋白質結構來決定其二級結構。
對生物大分子的二級結構含量可以以光譜來初步估計。對於蛋白質,最常用的方法是圓二色性(Circular dichroism), (利用長紫外線,波長範圍170-250nm)。在獲得的光譜吸收曲線上,α螺旋結構會在208nm及222nm兩處同時出現極小值,而204nm和207nm處出現單個極小值則分別表示存在無規捲曲和β折疊結構。另一個較常用的方法是紅外光譜,它可以偵測因氫鍵所造成胺基的震盪。而光譜中,測定二級結構最準確的方法是利用核磁共振光譜所紀錄的化學位移,由於儀器和樣品製備上的原因,這一方法較為少用。
類型
編輯蛋白質
編輯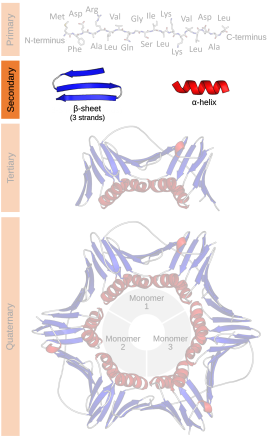
| 幾何屬性 | α-螺旋 | 310 螺旋 | π-螺旋 |
|---|---|---|---|
| 每一圈的殘基 | 3.6 | 3.0 | 4.4 |
| 每個殘基的翻譯 | 1.5 Å(0.15 nm) | 2.0 Å(0.20 nm) | 1.1 Å(0.11 nm) |
| 螺旋半徑 | 2.3 Å(0.23 nm) | 1.9 Å(0.19 nm) | 2.8 Å(0.28 nm) |
| 間距 | 5.4 Å(0.54 nm) | 6.0 Å(0.60 nm) | 4.8 Å(0.48 nm) |

蛋白質的二級結構包含局部殘基之間由氫鍵所調節的相互作用。最普遍的二級結構就是α螺旋及β折疊。經計算後發現其他螺旋,例如310螺旋及π螺旋,在能量上有着有利的氫鍵模式,但這些螺旋卻是在自然的蛋白質中是很稀有的,要α螺旋在中央進行不利的骨架包裝後,才可在末端中發現。緊的轉角、鬆開及靈活的環會連結更多「規則的」二級結構。任意形並非真正的二級結構,但卻是一類缺乏規則的二級結構的形態。
胺基酸在形成不同的二級結構上有着不同的能力。脯氨酸及甘胺酸會在轉角上出現,並且可以瓦解α螺旋骨架的規則形態,但兩者卻有着不正常的形態能力。在蛋白質內採用螺旋形態的胺基酸有蛋氨酸、丙氨酸、亮氨酸、穀氨酸及賴氨酸(胺基酸單字母編號為「MALEK」);相反,大型的芳香性殘基(色氨酸、酪氨酸及苯丙氨酸)及Cβ分枝的胺基酸(異亮氨酸、纈氨酸及蘇氨酸)則採用β折疊形態。但是,若單以序列來看,這些都不足以構成一個可靠的方法來預測二級結構。
DSSP編號
編輯DSSP是「Dictionary of Protein Secondary Structure」的縮寫,它是一編文章正式列出已知三維結構的蛋白質二級結構。DSSP編號一般是用單一英文字母來描述蛋白質二級結構。[3]二級結構是根據氫鍵模式來指定的。[4][5]
- G:3轉角螺旋(亦即310螺旋)。最短長度為3個殘基。
- H:4轉角螺旋(α螺旋)。最短長度為4個殘基。
- I:5轉角螺旋(π螺旋)。最短長度為5個殘基。
- T:氫鍵轉角(3、4或5個轉角)。
- E:平行的β折疊,或/及反平行的折疊形態(延伸鏈)。最短長度為2個殘基。
- B:獨立β橋內的殘基(一對β折疊氫鍵)
- S:彎曲(唯一非氫鍵的指定)
所有不是以上形態的殘基,在DSSP都是以空格來指定的,而有時則以C來代表捲曲或L來代表環。螺旋(即G、H及I)及折疊形態都需要一定的長度。這即是指兩個在一級結構鄰接的殘基必須形成相同的氫鍵模式。如果螺旋或折疊的氫鍵模式太短,就會分別以T或B來編碼。當中亦有其他蛋白質二級結構編號,但卻較少使用。
DSSP氫鍵定義
編輯由於二級結構是以氫鍵來定義,所以氫鍵的正確定義十分重要。DSSP內二級結構的標準氫鍵是一個純粹靜電模型。對於羰基的碳及氧,指定的電荷分別為:


而靜電能是:
![{\displaystyle E=q_{1}q_{2}\left[{\frac {1}{r_{ON}}}+{\frac {1}{r_{CH}}}-{\frac {1}{r_{OH}}}-{\frac {1}{r_{CN}}}\right]*332\ \mathrm {kcal/mol} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/a3d1e8288de134883df06a047e45cfd307537e48)
根據DSSP,一個氫鍵只有在E少於-0.5 kcal/mol才會存在。雖然上述的方程式都只是一個相對於氫鍵能量的估算,但都一般接受作為定義二級結構的工具。
蛋白質二級結構預測
編輯早期蛋白質二級結構預測的方法是建基於胺基酸形成螺旋或折疊的傾向,而有時須聯同估計形成二級結構的能量的方法來使用。這些方法在預測殘基的三種狀態(螺旋、折疊或捲曲)可以有約60%的準確性,若使用多重序列比對可以將準確性大幅提升至80%。多序列比對可以知道胺基酸在某一位置的完正分佈(包括在其附近的位置,一般在每一邊的7個殘基),而演化過程提供了結構趨向更明確的圖畫。例如,在蛋白質某位置的甘胺酸,本身已表明那是一個任意形。但是多序列對比可以發現,在接近十億年演化後95%的蛋白質中,那是一個有利螺旋的胺基酸。再者,若在那位置檢測平均疏水性,亦會發現其殘基可溶性是與α螺旋一致。綜合來說,這些因素顯示原先蛋白質內甘胺酸是α螺旋結構,而非任意形。多種方法都會結合已有的數據來組成三種狀態的預測,這些方法有神經網絡、隱馬爾可夫模型及支持向量機。現代預測方法亦可在每一個位置的預測結果提供信賴分數。
二級結構預測方法一直不斷地在校準,例如EVA實驗。基於約270個星期的測試,最準確的方法要算是PsiPRED(頁面存檔備份,存於互聯網檔案館)、SAM[永久失效連結]、PORTER、PROF及SABLE(頁面存檔備份,存於互聯網檔案館)。有趣的是,在這多種方法中找出共識或一致,並不能提升它們的準確性。最大改善的地方似乎是在β股的預測,因為所使用的方法會忽視一些β股段。整體上而言,最高的預測準確性只可以達90%,因DSSP的標準方法的性質,與校準的預測相違背。
準確的二級結構預測是三級結構預測的重要原素。例如一個確定的βαββαβ二級結構模式,就是鐵氧化還原蛋白的記號。
核酸
編輯核酸亦有二級結構,大部份都是單股核糖核酸[來源請求](RNA)分子。RNA二級結構可以分為螺旋(緊接的鹼基對)及不同種類的環(被螺旋圍繞的不成對核苷酸)。莖環結構是一個鹼基對螺旋結構,末端為短少的不成對環。這種莖環結構非常普遍,並且是建構大型結構模體,如三葉草結構(即如在轉運RNA中的四個螺旋結點)的基本單位。內環結構(在長鹼基對螺旋中的短而不成對鹼基)及膨出(在螺旋股中額外插入,但卻在相對股中沒有配對的鹼基)亦很經常會出現。最後,偽結及base triples亦會出現在RNA。
由於RNA二級結構差不多全都是由鹼基對作為中介,它可以說是確定在一個分子或複合物中哪些鹼基成對。但是,傳統的華生—克里克鹼基對並非唯一在RNA的配對方法,霍氏配對方法亦很普遍。

去氧核糖核酸(DNA)的二級結構主要是各種形式的螺旋,特別是B型雙螺旋、此外還有A型雙螺旋、Z型雙螺旋、三螺旋和四螺旋結構等[6]。除了上述3種最常見的標準二級結構(B型、A/C型和Z型)外,細胞內DNA在特殊條件下亦可形成其他幾種非標準二級結構,如彎曲(bending)、十字形(cruciforms)、三螺旋(triple helix)、滑動(slipped mispaired DNA,SMP-DNA)錯配和剪輯翻轉(base flipping)等[6]。
RNA二級結構預測
編輯生物信息學的其中一種應用是使用預測的RNA二級結構來搜尋用作RNA功能形式而非編碼的基因組。舉例來說,小分子RNA有着由小內環中斷的長莖環結構。計算可能的RNA二級結構可以用動態規劃方法,但是它不能偵測出偽結或是其他鹼基對沒有全面網羅的情況較通用的方法有隨機上下文無關語法。Mfold是一個使用動態規劃的網站。
在很多RNA分子,二級結構對RNA正常功能非常重要,有時甚至於較序列重要。這可以幫助用於分析非編碼RNA。RNA二級結構可以用電腦來提升預測準確性。[7],而其他生物信息學的應用會使用一些二級結構的概念來分析RNA。
應用
編輯蛋白質及RNA二級結構都可以用在協助多重序列比對。這種比對在加入有關的二級結構資料後,可以變得更為準確。但有時對RNA卻不太有用,這是由於RNA鹼基對比序列更受到高度保存。一些不能比對一級結構的蛋白質,二級結構有時亦可以找出它們之間的關係來。
參考文獻
編輯- ^ C Branden; J Tooze. Introduction to Protein Structure 2nd ed. New York: Garland Publishing. 1999.
- ^ Steven Bottomley. Interactive Protein Structure Tutorial. 2004 [January 9, 2011]. (原始內容存檔於2010-12-19). (頁面存檔備份,存於互聯網檔案館)
- ^ Kabsch W; Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983, 22: 2577–2637. PMID 6667333.
- ^ L. Pauling; R.B Corey. Configurations of polypeptide chains with favored orientations of the polypeptide around single bonds: Two pleated sheets. Proc. Natl. Acad. Sci. Wash. 1951, 37: 729–740.
- ^ L. Pauling; R.B. Corey and H.R. Branson. Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. Wash. 1951, 37: 205–211.
- ^ 6.0 6.1 楊榮武. 第六章第三节:核酸的高级结构. 生物化学原理 2. 北京: 高等教育出版社. 2012. ISBN 978-7-04-035696-0. OCLC 910676076.
- ^ M. Zuker. Computer prediction of RNA structure. Methods in Enzymology. 1989, 180: 262–88.
延伸閱讀
編輯- Branden C, Author J. Introduction to protein structure 2nd. New York: Garland Science. 1999. ISBN 978-0815323051.
- Pauling L, Corey RB. Configurations of Polypeptide Chains With Favored Orientations Around Single Bonds: Two New Pleated Sheets. Proc. Natl. Acad. Sci. U.S.A. 1951, 37 (11): 729–40. PMC 1063460
 . PMID 16578412. doi:10.1073/pnas.37.11.729. (The original beta-sheet conformation article.)
. PMID 16578412. doi:10.1073/pnas.37.11.729. (The original beta-sheet conformation article.) - Pauling L, Corey RB, Branson HR. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. U.S.A. 1951, 37 (4): 205–11. PMC 1063337 . PMID 14816373. doi:10.1073/pnas.37.4.205. (alpha- and pi-helix conformations, since they predicted that helices would not be possible.)

參見
編輯外部連結
編輯- NetSurfP – Secondary Structure and Surface Accessibility predictor(頁面存檔備份,存於互聯網檔案館)
- PROF(頁面存檔備份,存於互聯網檔案館)
- ScrewFit
- PSSpred(頁面存檔備份,存於互聯網檔案館) A multiple neural network training program for protein secondary structure prediction
- Genesilico metaserver Metaserver which allows to run over 20 different secondary structure predictors by one click
- SST(頁面存檔備份,存於互聯網檔案館) webserver: An information-theoretic (compression-based) secondary structural assignment.